海河文库|重磅!FDA 发布 510 (k) 转让指导原则草案,医疗器械企业必看

近期,FDA发布了510(k)转让指导原则草案,明确医疗器械企业转让510(k)时的责任与流程,对已获批的510(k)在转让时的常见疑问进行了澄清。此指导原则核心想表达的是:当已获批510(k)的医疗器械,其持有人(公司或个人)发生变更,例如由于公司收购、产品授权、法人变更等情况,就构成510(k)转让。此时不一定需要重新提交510(k),但新旧持有人都必须履行一系列合规责任。下面让我们看一下具体问题的Q&A:
Q1: 谁需要提交510(k)?

A: a.国内生产商首次将器械引入美国商业分销时必须提交510(k);
b.再制造商(Remanufacturer);
c.一次性使用器械的再处理商(Reprocessor of single-use devices);
d.规格开发者(Specification developer)
e.外国生产商(Foreign manufacturer)
若器械正在销售中,或者重新将其投入市场,且该器械即将在设计、组件、制造方法或预期用途方面发生重大变化或修改,需要提交新的510(k)申请;若器械无实质性变更,且新持有人仅延续现有商业分销(非“首次引入市场”),则无需重新提交。
Q2: 同一器械能否存在多个510(k)持有人?
A: 不能,FDA规定同一时间仅允许1个持有人。
Q3: 哪些角色需要使用K号进行器械列示?
A:合同制造商、合同灭菌商、重新包装商(Repackager)、贴标商(Relabeler)这些角色都应使用510(k) K号进行列示。
Q4:受让510(k)后是否需要重新提交?
A: 通常不需要,但需满足:
•器械未发生重大变更(设计/组件/制造方法/预期用途不变);
•新持有人不是首次将该器械引入美国市场,而是延续商业分销。
需重交情况:若转让后对器械进行重大变更。
Q5: 转让后新持有人有哪些义务?
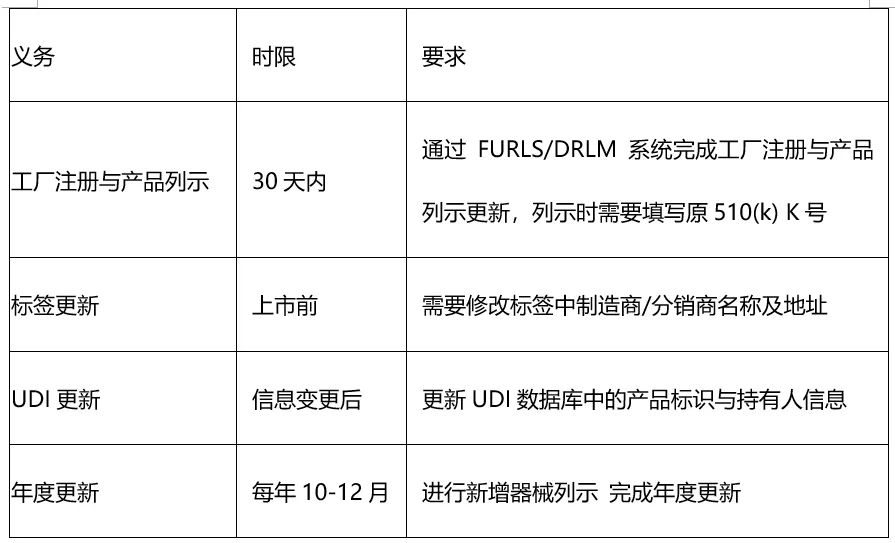
请注意,如未履行上述义务将导致器械被认定为掺假产品或者冒牌产品,这可能导致产品被拒绝进口、召回,甚至罚款。
原持有人义务:
•提供完整的510(k)文件资料、测试数据、变更记录等;
•若终止生产或不再负责该器械,需在FDA注册系统中更新器械列表,删除被转让器械的列示信息;
•合理配合新持有人的注册与上市工作。
欢迎后台留言“510 (k) 转让”咨询小编索要原文件。
海河生物可以提供的服务
FDA 发布 510 (k) 转让指导原则草案,旨在规范 510 (k) 许可转让流程、责任划分及合规要求等。海河生物作为专业为医疗器械和药品相关机构提供产品全生命周期服务的平台性公司,目前已完成超 3000 个美国 FDA 510(k)申请,特别是为中国医疗器械所有细分领域的“首台套”产品进入欧美市场提供了服务。海河生物可根据客户需求提供法规咨询、注册认证辅导、检测服务、质量体系等相关服务。在中国医疗器械产品逐浪出海的宏伟航程中,海河生物了解中国企业在注册申报中的痛点和难点,若您的器械出海美国市场需求,速邮件sales@mid-link.net 联系我们。
如需转载,请标明来自海河生物
海河生物是一家专业为医疗器械、药品以及药械结合产品相关的研究机构、研发和生产企业以及相关监管部门提供产品全生命周期服务的平台性公司,在生物医药CRO领域具有极高的行业地位。海河生物旗下业务涵盖产品全生命周期服务、服务型制造、智能制造和第三方检测认证,与我国“十四五规划”高度契合。
从事医疗器械、药品、药械结合产品和生物医药环境标准化及定制检测和监测服务,具有具有国内、国际先进检测标准相关资质以及国际实验动物饲养管理评估认证委员会 AAALAC 认证。
向客户提供中国、美国、欧盟、巴西、加拿大和东南亚国家等国家地区的医疗器械、药品和药械结合产品全生命周期法规政策咨询、注册认证辅导、上市后不良事件处理、技术转化、培训等相关服务。
海河生物的医疗器械委托研发生产平台,是按照国家药监局医疗器械生产质量管理规范、美国药监局 QSR 和 ISO13485 要求建立的为器械产学研、医工转化和初创型团队服务的平台,承接医疗器械产品定制、外包研发、外包生产的业务,生产能力涵盖 IVD 诊断试剂,IVD 诊断设备,医疗设备、外科高值耗材等产品 。
