FDA 510 (k) 数据库 6 大新增功能:器械出海的智能导航系统升级

FDA 510 (k) 数据库是美国食品药品监督管理局(FDA)面向公众开放的医疗器械检索平台,收录已获批上市的 510 (k) 产品信息。企业可通过该数据库查询辅助注册申报;公众可获取产品技术资料、审批状态等公开信息。
核心功能与查询入口

查询入口:https://www.accessdata.fda.gov/scripts/cdrh/cfdocs/cfpmn/pmn.cfm。
检索逻辑:
支持关键词、产品代码、企业名称、510 (k) 编号等多维度筛选,新增 6 个细分选项可与基础条件联用。
新增6大查询选项功能
目前,FDA 510(k) 数据库的查询页面新增了 6 个查询选项(见图 1)。接下来,我们将介绍如何使用这些新增的查询功能。
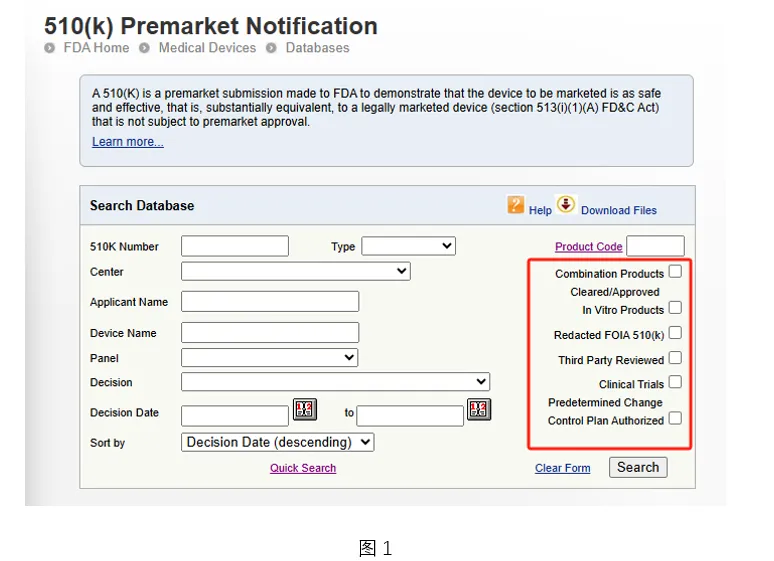
此选项用于检索所有被归类为组合产品的器械。目前通过此选项检索到的组合产品,均属于以器械为主的药械组合产品。
✅ 已获批体外诊断产品 (Cleared/Approved In Vitro Product)
与欧盟将医疗器械(MD)与体外诊断产品(IVD)分别立法监管不同,FDA 未对IVD 单独立法。因此,只要能够通过实质等同性证明其安全性和有效性,IVD 即可与医疗器械一样通过 510(k) 途径上市。值得注意的是,医疗器械与 IVD 的 510(k) 注册号在编号格式上并无区分标识。
此次新增的 IVD 查询选项,旨在方便用户直接检索 IVD 相关的 510(k) 公开文件。
✅ 依据FIOA公布510(k)文件 (Redacted FIOA 510(k))
FOIA 是《信息自由法案》(Freedom of Information Act) 的简称。公众依据该法案可向 FDA 提交申请,请求获取其持有但尚未主动公开的文件或记录。公众可依据 FOIA 向 FDA 申请公开特定产品的 510(k) 申请资料。FDA 收到 FOIA 申请后,需完成内部审查并与该 510(k) 资料的所有者进行沟通协调。同时,FOIA 申请人还需支付相应费用。因此,提交 FOIA 申请涉及一定的时间和经济成本。
“依据FIOA已公布的510(k)文件” 指的就是那些已被他人依据 FOIA 申请公开、且 FDA 已将对应的,经过处理的510(k) 资料提供给申请人的项目。通过此新增查询选项,公众可实时检索并免费下载这些已公布的510(k)文件。
重要提示:“已公布的510(k)” 文件中的大量关键信息通常经过修订处理(被遮挡或涂抹)。这是符合 FOIA 法案的要求的-510(k) 资料的所有者有权识别并对其认为敏感的、不应公开的商业信息进行修订。
✅ 第三方审核 (Third Party Reviewed)
FDA 监管范围广泛,涵盖食品、药品、医疗器械、化妆品、血液制品及烟草等。鉴于审评资源有限,FDA 授权符合条件的第三方审核机构对特定类别的医疗器械进行技术评估。通过此新增查询选项,公众可快速确认哪些产品支持通过第三方审核机构提交注册申请。
重要提示:
(1) 第三方审核仅适用于少数技术成熟、中低风险等级的医疗器械。
(2)该机制旨在补充 FDA 的审评资源,提高审评效率。
(3) 第三方审核机构仅拥有技术评估权,最终的 510(k) 审批决策权仍归属 FDA。
✅ 临床试验 (Clinical Trial)
公众可以通过这个新增查询直接筛查出带有临床试验数据的510(k)注册产品,并且通过此新增查询选项,公众可直接筛选出包含临床试验数据的 510(k) 获批产品。 筛选结果页面已关联ClinicalTrials.gov 数据库链接,用户可便捷访问并查看相关临床试验详细信息。
✅ 已授权预定变更控制计划 (Predetermined Change Control Plan Authorized)
FDA 的 预定变更控制计划 (Predetermined Change Control Plan, PCCP) 是一个相对新兴且至关重要的监管框架,适用于预期将进行计划内迭代更新的复杂医疗器械,尤其是在人工智能/机器学习 (AI/ML) 驱动的医疗器械领域。其核心目标在于解决一个关键矛盾:如何在确保医疗器械安全有效性的前提下,允许已获市场授权的 AI/ML 器械进行必要且持续的改进与更新。
若某产品的 510(k) 申请文件中包含 PCCP 并获得 FDA 授权,则意味着该器械后续在实施 PCCP 中预先批准的变更时,仅需按规定提交相应报告,而无需为每次变更单独提交新的 510(k) 申请。 通过此新增查询选项,公众可直接筛选出哪些已获批的 510(k) 产品获得了 PCCP 执行授权。
新增查询选择可与其他查询选项联用
公众可将新增的查询选项作为附加筛选条件,与其他查询条件组合使用,以获得更精准的搜索结果。
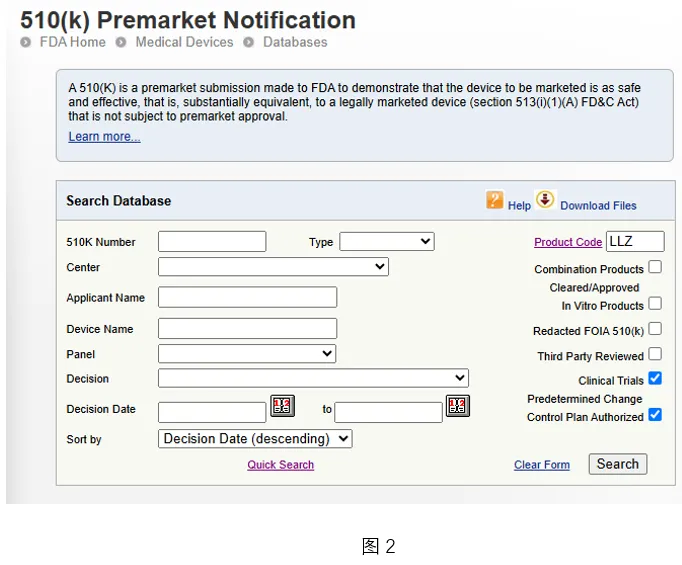
如图 2,我们可以在产品代码 LLZ 下,额外叠加应用“临床试验”、“已公布的510(k)文件”以及“已授权的预定变更控制计划”这三个筛选条件。如此,即可高效定位那些包含临床试验数据、可获取公开 510(k) 注册文件、且已获 PCCP 授权的影像后处理产品。
海河生物可以提供的服务
中国医疗器械正迎来全球化黄金期,海河生物目前已完成超 3000 个美国 FDA 510(k)申请,特别是为中国医疗器械所有细分领域的“首台套”进入欧美市场提供了咨询服务。在中国医疗器械产品逐浪出海的宏伟航程中,海河生物了解中国企业在注册申报中的痛点和难点,若您的器械出海美国市场需求,速邮件sales@mid-link.net 联系我们。
如需转载,请标明来自海河生物
